Access to these tools is limited to the Labex Cemeb users who must request an account via this link.
Other users can download workflows source codes from the git repository.
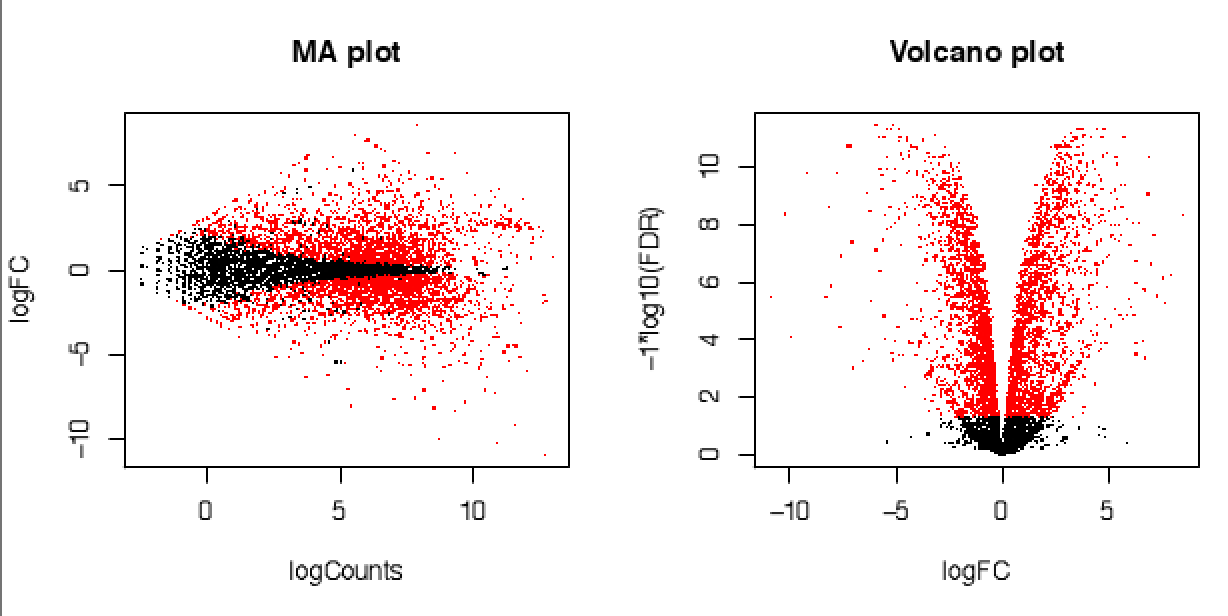
MBB RNAseq
Rnaseq reads can be cleaned and/or trimmed before quantifying abundances of transcripts (with Kallisto or Salmon). Differentially expressed genes can then be checked with edgeR or deseq2.
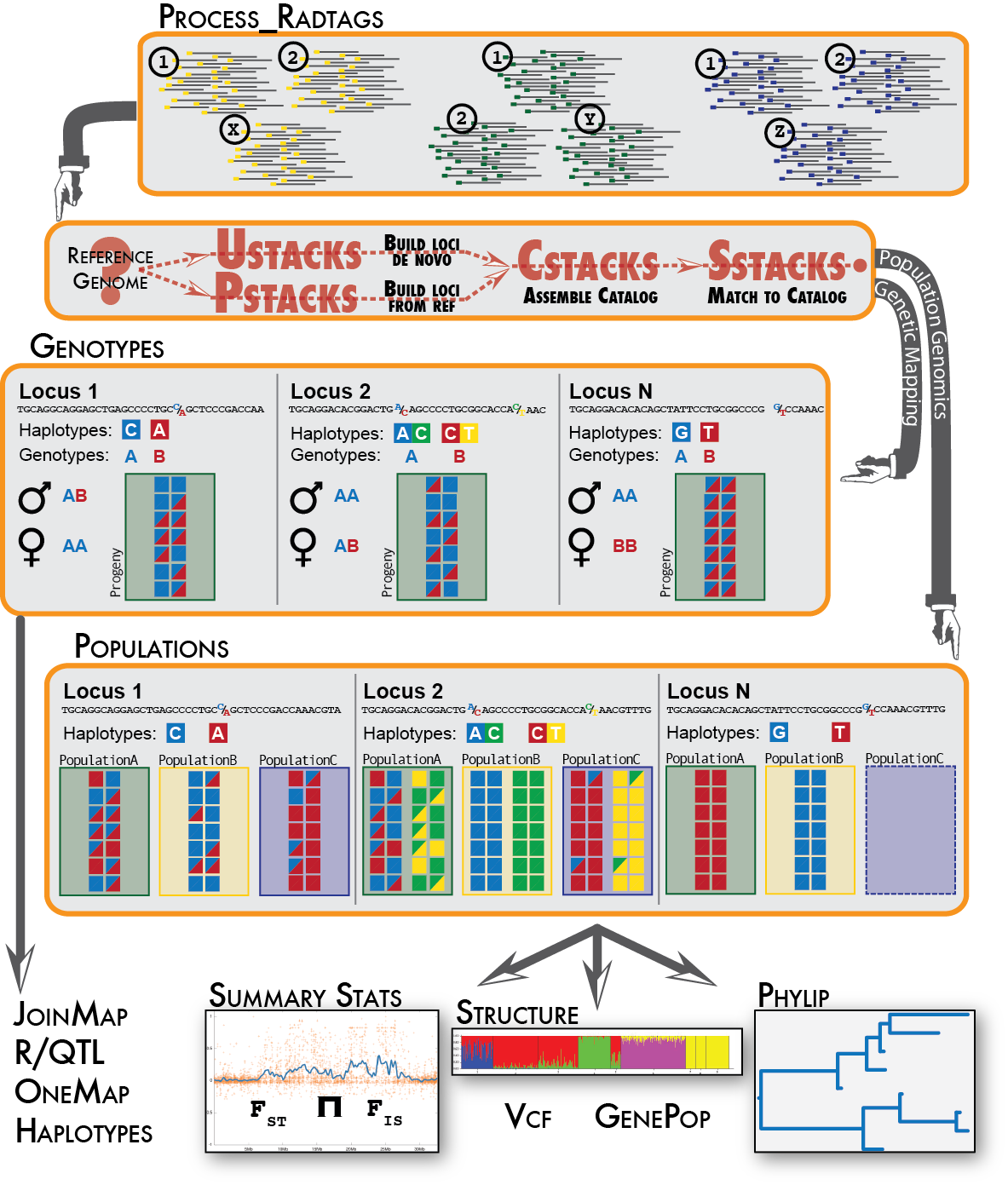
MBB RADseq on Reference
After quality checking of restriction enzyme-based data, such as RAD-seq, the sequences are demultiplexed and mapped with bowtie or bwa to a ref. genome. The resulting bam files are then analysed with gstacks and populations from the Stacks pipeline.
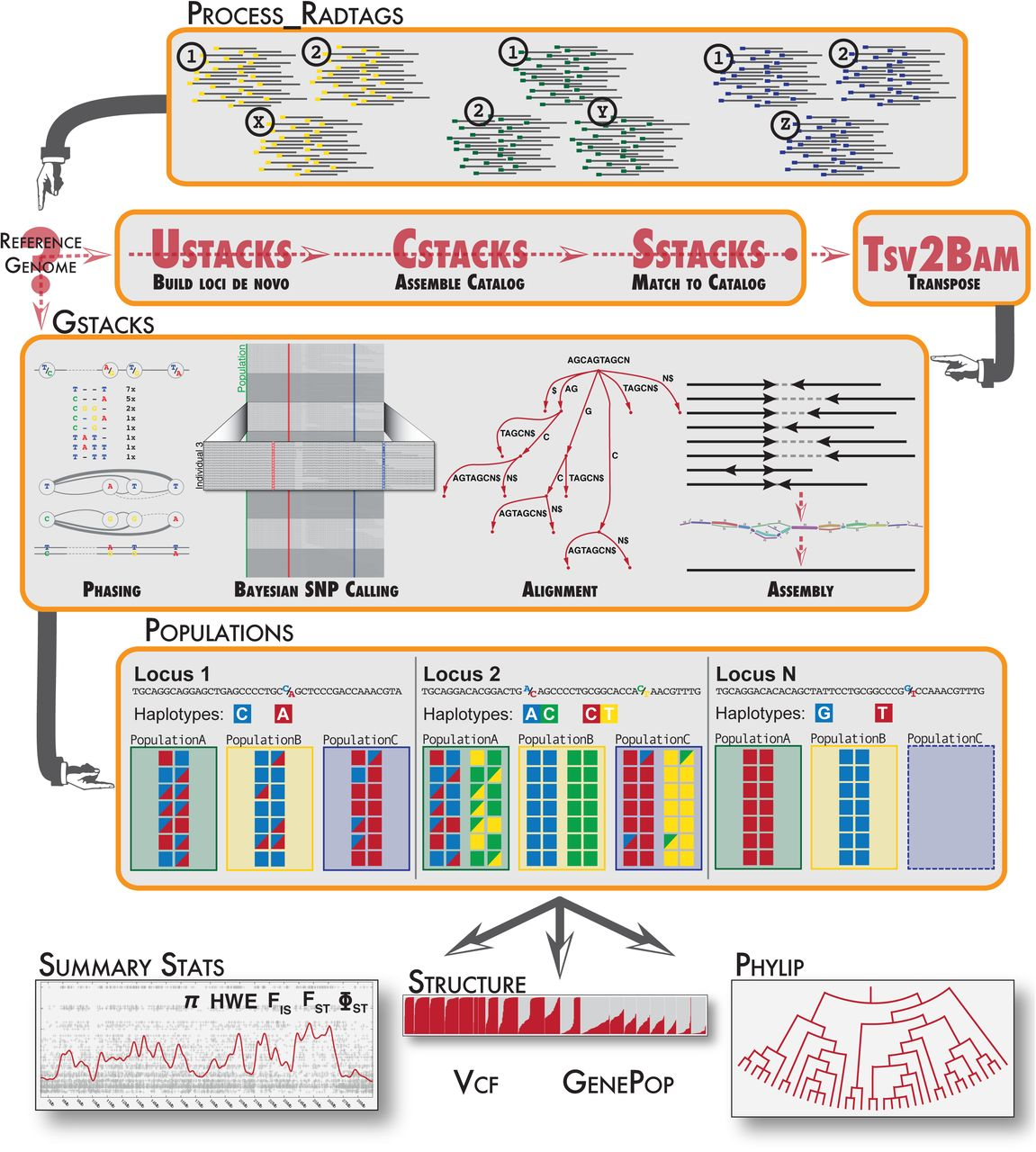
MBB denovo RADseq using Stacks
This tool will run Stacks components to build loci de novo, create catalog across the population and call snp and generate population-level summary statistics .
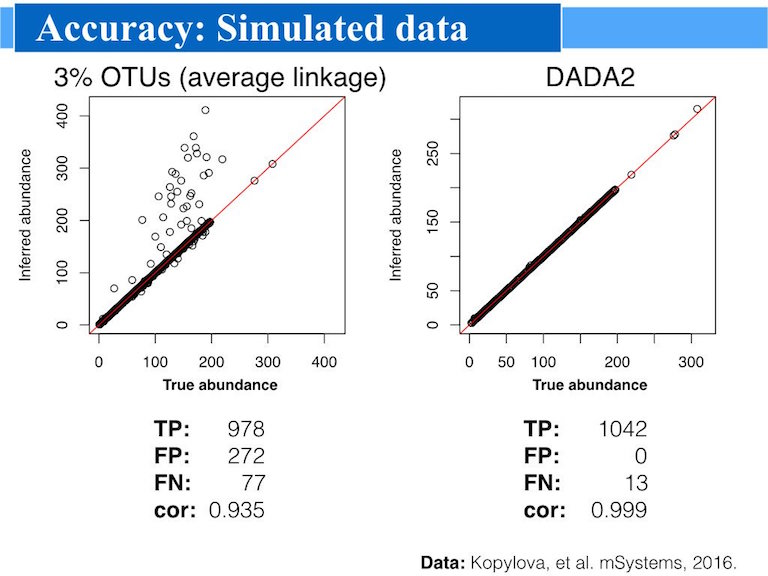
meta-barcode analysis with DADA2
Amplicon data (16S, 18S or ITS) are analysed with a dada2 typical workflow ":" filtering and trimming reads, learning errors rates, constructing amplicon sequence variant table ASV table and assigning taxonomy from Silva, rdp and GreenGene databases.
Assemble shotgun seq with Megahit and annotate mitochondrion with MitoZ.
Shotgun reads can be quality checked before a fast and memory-efficient NGS assembling with Megahit. The mitochondrial contigs are then find and annotated with Mitoz procedures. A Circos and coverage plot and then produced in a fastqc html report.
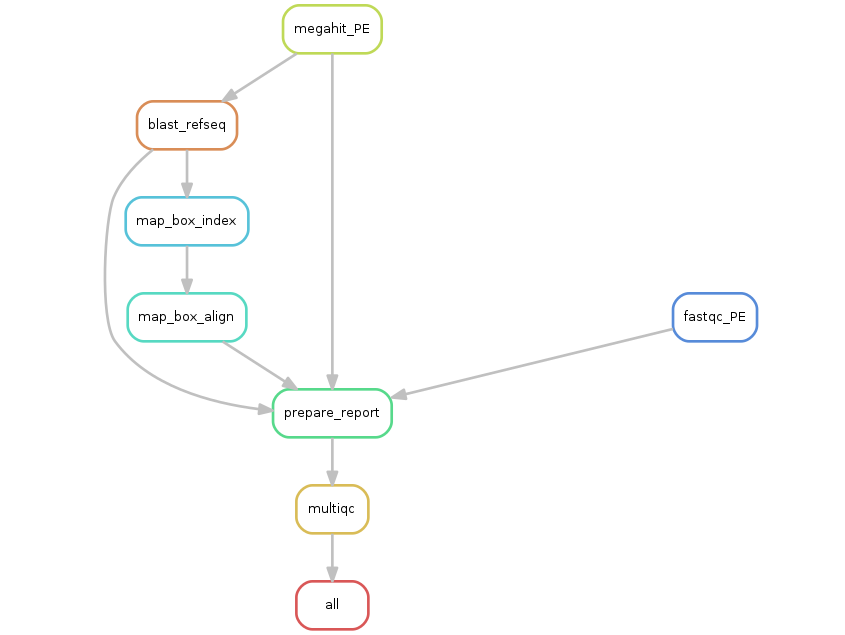
Assemble shotgun seq with Megahit and blast on viral Refseq .
Shotgun reads can be quality checked before a fast and memory-efficient NGS assembling with Megahit. The final contigs are blasted against RefSeq viral database to detect and extract known virus sequences.
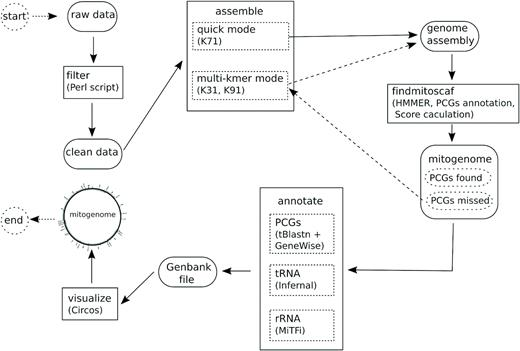
Assemble shotgun seq with MitoZ and annotate mitochondrion .
Based on MitoZ workflow, this pipeline assemble genome, search for mitogenome sequences from assembly, annotate mitogenome, and produce a mitogenome visualization.

Map reads to a reference and call variants.
Reads can be preprocessing with fastp tool before mapping on your reference genome (with Bwa or bowtie). The mapping result can be followed with mark duplicates, Indel realignment before variant calling with Gatk haplotype caller or mpileup bcftools.
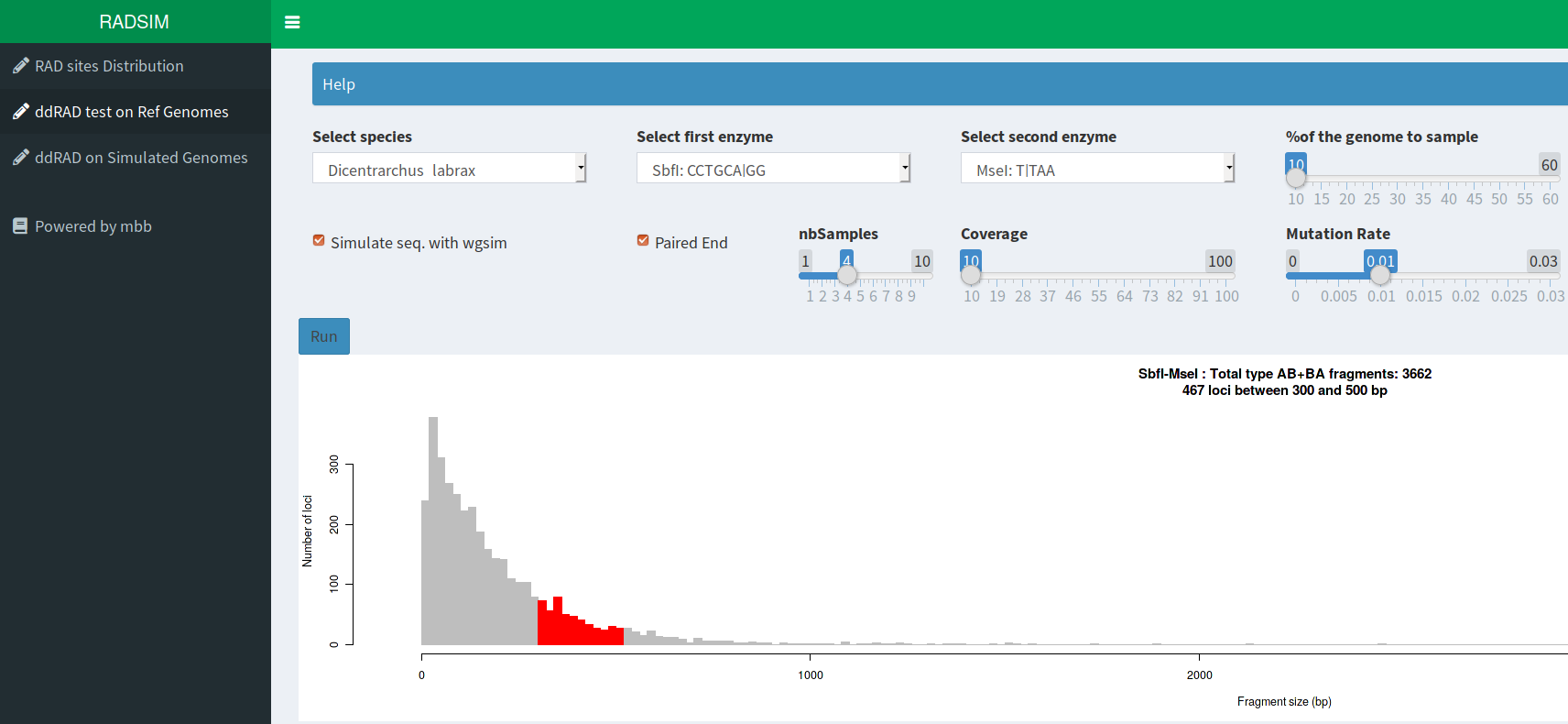
RAD simulator
This tool will sample a % of contigs or chromomosmes from selected ref. genome. It then cut those chr with a pair of selected enzymes and plot the distrib. of expected fragments sizes. A simulation with wgsim can sequence reads (with a selected size in single or paired-end mode). To mimic divegence from the ref. genome this step can be preceded by a procedure applying errors, substitutions and indels to the selected fragments. The obtained reads are then mapped and snp called to the ref. genome
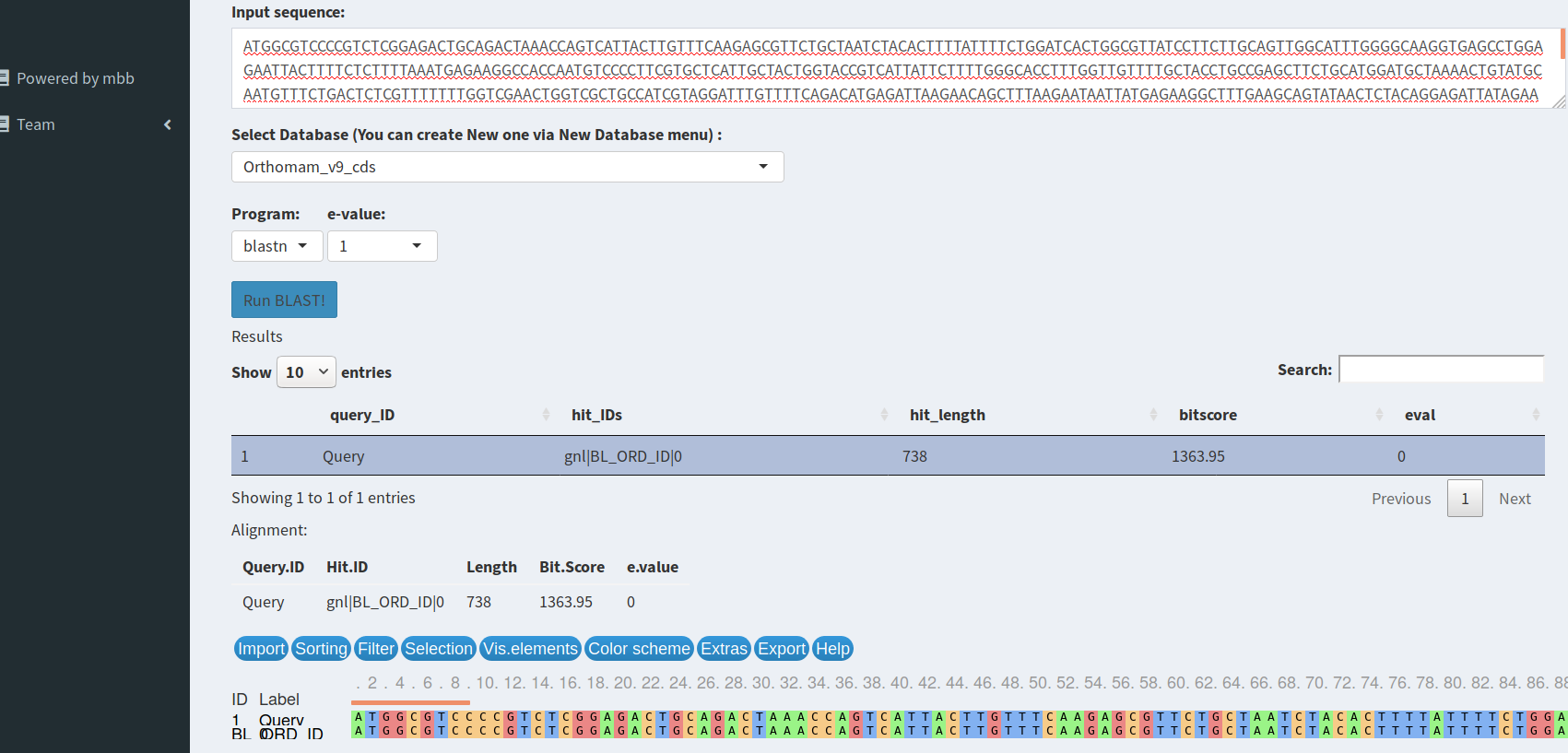
A shiny Interface to NCBI Blast
With this tool you can either blast your sequences on NCBI nr/nt database or on your own blast database. Best Hits results are tabulated and alignment are plotted with msaR.
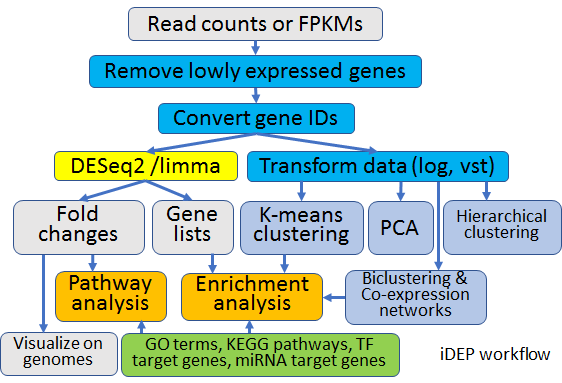
Integrated Differential Expression and Pathway analysis iDEP
This is an instance of The integrated web application for differential expression and pathway analysis iDEP (https://www.biorxiv.org/content/10.1101/148411v1)
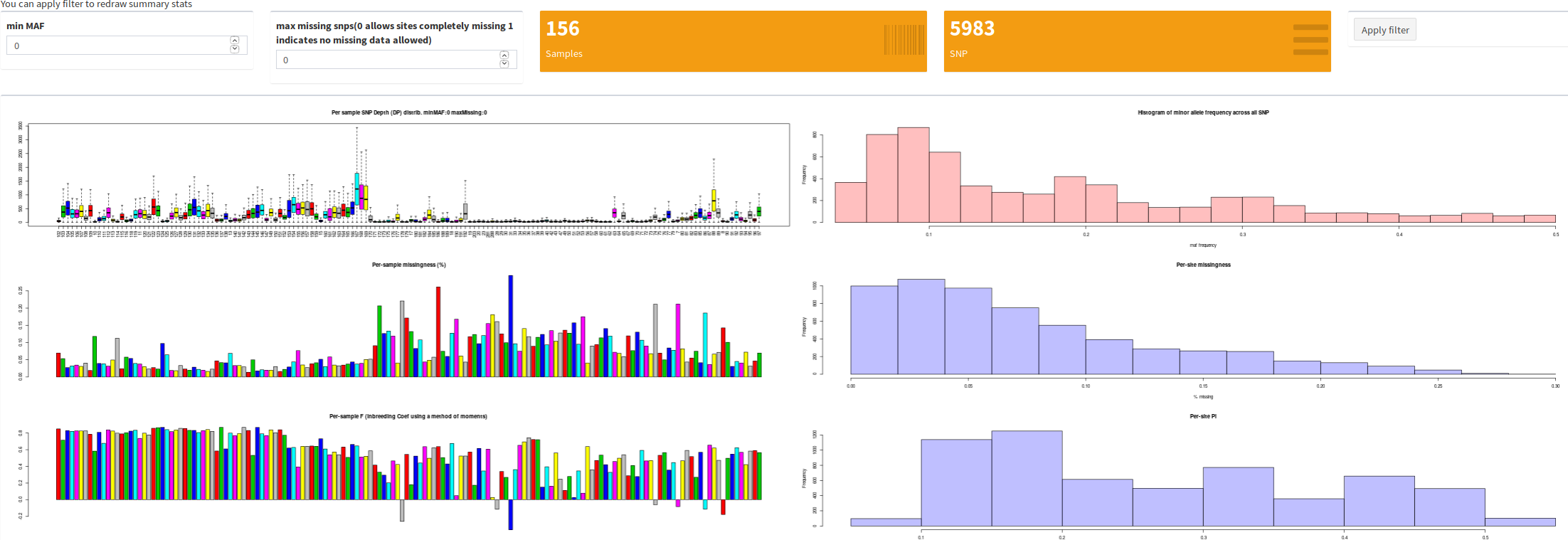
MBB ShinyVCFAnalyser
This interface can help analyse a medium sizes multi-individual vcf file as produced by stacks. Different summary statistics are plotted. PCA, Fst, IBS and fastStructure are also implemented.

|
|
|
|
|
|
|